重磅!2021年中国大输液行业相关政策汇总及解读(全) 全面实行MAH制度
注射给药途径虽然有吸收快、药量准确可控的优点,但可引起组织损伤、疼痛、感染,甚至引发严重不良反应的缺点也十分明显。在经历了三十多年的爆发式发展之后,我国大输液安全事故的频发逐渐引起监管部门的关注,相关行业政策也逐渐趋严。
2019年,国家药监局发布关于贯彻实施《中华人民共和国药品管理法》有关事项的公告,自2019年12月1日起,取消药品GMP、GSP认证,不再受理GMP、GSP认证申请,不再发放药品GMP、GSP证书。
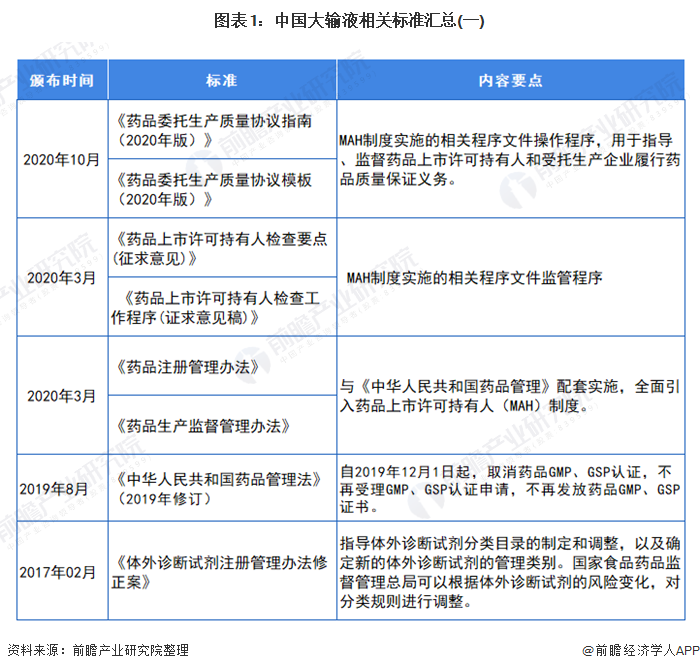
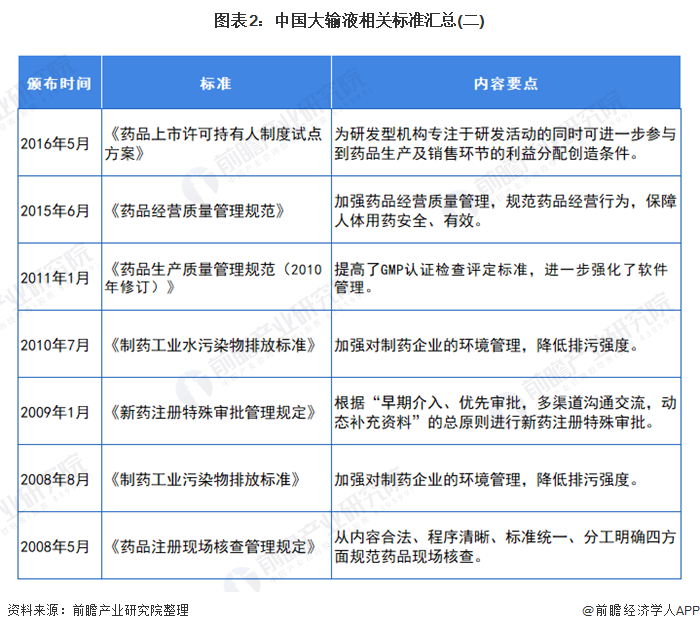
行业相关标准
我国制定了严格的行业标准来规范医药行业,其中主要有:《药品管理法》(中华人民共和国主席令第45号)、《药品管理法实施条例》(国务院令第360号)、《药品注册管理办法》(国家药监局令第28号)、《药品经营许可证管理办法》(国家药监局令第6号)、《药物临床试验质量管理规范》(国家药监局令第3号)、《药品进口管理办法》(国家药监局令第4号)、《药物非临床研究质量管理规范》(国家药监局令第2号)、《药品经营质量管理规范》(国家药品监督管理局令第20号)、《药品生产质量管理规范》(国家药品监督管理局令第9号)等。与化学药品制剂行业发展相关的法律法规主要如下表所示:
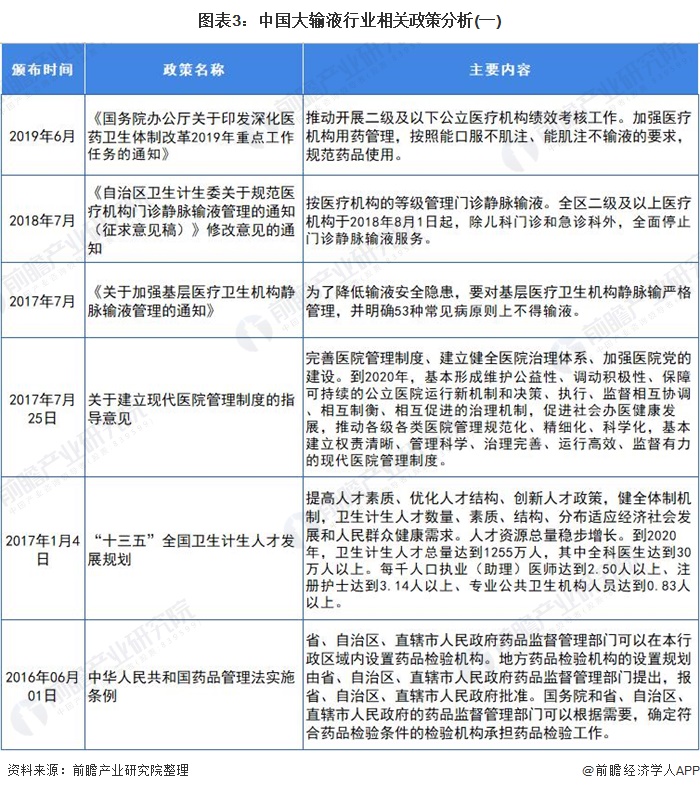
行业相关政策
注射给药途径虽然有吸收快、药量准确可控的优点,但可引起组织损伤、疼痛、感染,甚至引发严重不良反应的缺点也十分明显。在经历了三十多年的爆发式发展之后,我国大输液安全事故的频发逐渐引起监管部门的关注,相关行业政策也逐渐趋严。
2012年8月,有“史上最严限抗令”之称的《抗菌药物临床应用管理办法》正式实施。根据《办法》,抗菌药物将依据安全性、疗效、细菌耐药性、价格等因素,被分为非限制使用级、限制使用级和特殊使用级三个级别。医疗机构和医疗人员应严格掌握使用抗菌药物预防感染的指征。违规使用抗菌药物的医师,将被警告、限制处方权、吊销执业证书,甚至追究刑事责任。
据国家药品不良反应监测中心发布的2019年药品不良反应/事件报告数据显示,注射给药占62.8%,远超占比32.5%的口服给药,在注射给药中,静脉注射给药又占到了92.5%。报告再次强调:能口服给药的,不选用注射给药;能肌肉注射给药的,不选用静脉注射或滴注给药。预计近几年,我国大输液行业政策仍将保持高压状态。
行业最新动态解读
1、取消GMP、GSP认证
2012年,我国拥有300多家大输液企业,在“限抗令”的推动下,实力较弱的小型企业退出市场速度加快。到2013年底,有110余家大输液企业由于未能通过GMP认证停产,行业企业数量迅速减少了三分之一左右。截止2021年4月,在企查查输入“大输液”关键字,选出经营范围为大输液的、存续/在业的制造业企业数量不足100家。我国大输液行业经过不断的优胜劣汰,许多企业被淘汰,行业集中度大幅提升。
2019年,国家药监局发布关于贯彻实施《中华人民共和国药品管理法》有关事项的公告,自2019年12月1日起,取消药品GMP、GSP认证,不再受理GMP、GSP认证申请,不再发放药品GMP、GSP证书。2019年12月1日以前受理的认证申请,按照原药品GMP、GSP认证有关规定办理。
取消药品GMP、GSP认证,有着如下意义:
1、取消GMP认证并不会降低药品质量标准,也不意味着药企生产门槛的降低,相反,药企将面临更加常态化和严苛的检查。以往药品GMP认证相当于颁给企业一个五年有效的合格证,即使企业不按照规范生产,也往往因有政府认证的资质而规避自身责任。未来取消以事前认证形式监管之后,药企将面临更加严格的各类检查,特别是事先不告知的飞行检查。
2、随着产业管理水平、质量意识的提高,简化行政审批,进行简政放权,将极大地降低了企业运营成本,有效激发医药产业活力,利于药品安全与行业创新。
3、取消GMP、GSP认证和落实药品上市许可持有人(MAH)制度之间是相互促进的。药品上市许可人制度是指将上市许可与生产许可分离的管理模式。这种机制下,上市许可和生产许可相互独立,上市许可持有人可以将产品委托给不同的生产商生产,药品的安全性、有效性和质量可控性均由上市许可人对公众负责。M
AH制度是国际较为通行的药品上市、审批制度,是一项与世界接轨的制度,具有一定的制度优势,可在一定程度上缓解目前“捆绑”管理模式下出现的问题,从源头上抑制制药企业的低水平重复建设,提高新药研发的积极性,促进委托生产的繁荣,从而推进我国医药产业的快速发展。
也就是说,以往药品在申报前就和GMP认证的药厂绑定了,认证的取消使药企可先申报新药后,再委托任何一个符合条件的药厂去加工生产,这对于鼓励药品的研发生产具有促进意义。
4、从行政审批制度改革的深入程度来看,一整套鼓励药品研发、创新的配套制度正在逐渐完善。
2、全面实行MAH制度
2020年3月,为进一步规范持有人检查工作,国家药监局组织起草了《药品上市许可持有人检查工作程序(征求意见稿)》《药品上市许可持有人检查要点(征求意见稿)》。文件指出,持有人检查包括现场检查和书面检查两种形式,现场检查指药品监督管理部门委派检查组至持有人研制、生产、经营相关活动现场进行核查的检查方式;书面检查指药品监督管理部门要求持有人递交检查材料,并对其递交材料进行核查的检查方式。
同样是2020年3月,国家市场监督管理总局公布《药品注册管理办法》和新修订的《药品生产监督管理办法》,两部规章将于2020年7月1日起正式施行,与《中华人民共和国药品管理》配套实施。规章中提到,全面落实药品上市许可持有人制度。
明确申请人为能够承担相应责任的企业或者药品研制机构等,要求建立药品质量保证体系,对药品的全生命周期进行管理,开展上市后研究,承担上市药品的安全有效和质量责任。
《药品注册管理办法》和《药品生产监督管理办法》是药品监管领域的两项核心配套规章,分别是我国药品研发和注册管理和上市药品生产管理的重要操作性规章,也是继《疫苗管理法》于2019年6月出台、《药品管理法》于2019年8月完成重大修订后,药品监管领域后续出台的两个重量级部门规章,这两个规章将分别替代目前施行的2007年《药品注册管理办法》和2004年《药品生产监督管理办法》。
新《药品管理法》的最大变化也是最大亮点就是药品上市许可持有人(MAH)制度的全面引入(实际上早于新《药品管理法》问世的《疫苗管理法》已正式使用了这一试点多年的创新制度),药品上市许可持有人作为药品行业监管的核心对象和主要责任载体贯穿了整部新法,药品上市许可持有人也将是“药品注册证书”中须载明的一项关键信息。
新法要求持有人应具备质量管理能力、风险防控能力和责任赔偿能力,对药品的非临床研究、临床试验、生产、经营、上市后研究、不良反应监测及报告与处理等承担责任,依法对药品研制、生产、经营、使用全过程中药品的安全性、有效性和质量可控性负责。
2020年10月,为贯彻《药品管理法》有关规定,进一步加强药品生产监督管理,国家药监局组织制定了《药品委托生产质量协议指南(2020年版)》和《药品委托生产质量协议模板(2020年版)》,用于指导、监督药品上市许可持有人和受托生产企业履行药品质量保证义务,自发布之日起施行。
由此可见,自2019年取消药品GMP、GSP认证后,国家进一步出台了一系列的规章制度,促进MAH制度的全面实施。
3、GMP证书尚在有效期范围内的部分大输液企业
自2019年,国家药监局取消药品GMP、GSP认证后,国家不再受理GMP、GSP认证申请,不再发放药品GMP、GSP证书。因此,持有GMP认证的大输液企业越来越少。截止2021年4月,前瞻统计的GMP证书尚在有效期范围内的部分企业和证书如下:

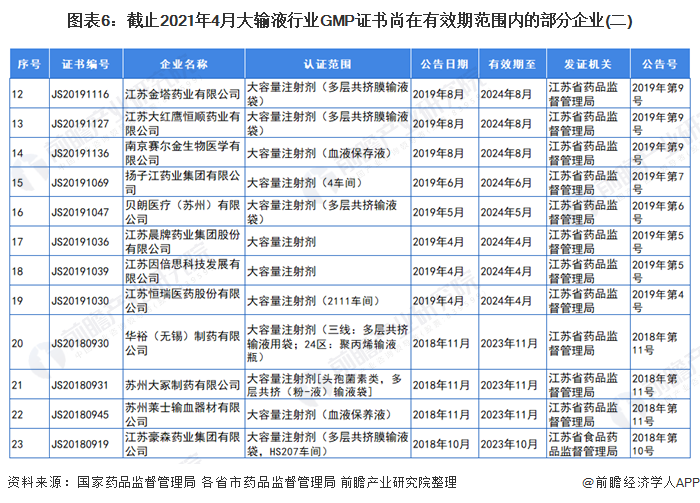
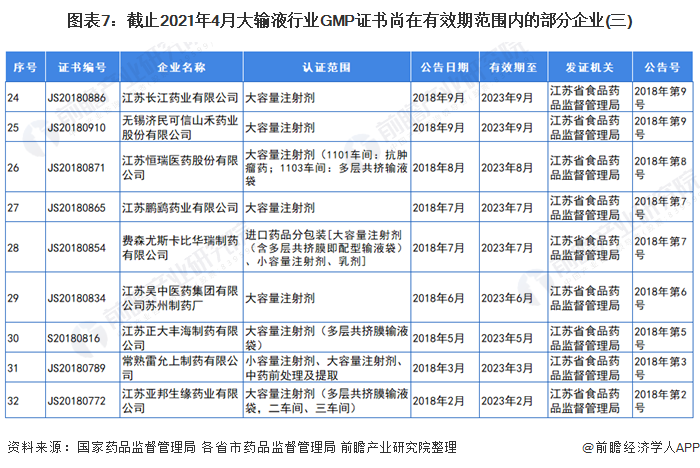
注:证书编号CN开头的为国家药品监督管理局颁发的证书,有效期范围内的企业已全部展示。其他证书编号开头的为各省市药品监督管理局颁发的证书,以北京市和江苏省为例。
(文章来源:前瞻产业研究院)
声明:文章仅代表原作者观点,不代表本站立场;如有侵权、违规,可直接反馈本站,我们将会作修改或删除处理。



















